ian_j_wilson@outlook.com
Google Scholar Profile
| September 2023 | SYNOD Software in the paper
Copy number analysis from genome sequencing data of 11,754 rare disease parent-child trios: a model for identifying autosomal recessive human gene knockouts including a novel gene for autosomal recessive retinopathy. E Olinger, IJ Wilson, S Orr, M Barroso-Gil, R Neatu et al. - Genetics in Medicine Open, 2024 link to paper |
| May 2017 | Diversit-tag. Code for the paper Abundant and equipotent founder cells establish and maintain acute lymphoblastic leukaemia by A Elder, S Bomken, I Wilson et al.. Leukemia volume 31: 2577–2586 is on github at github.com/ijwilson/diversit-tag. |
| January 2017 | JAGS Analysis of Copy Number A Second document Estimating Copy Number Frequencies from Censored Genotype Data looks at the differences between inference with all genotypes and for censored data. |
| January 2016 |
Mitochondrial DNA sequence characteristics modulate the size of the genetic bottleneck 2016. Wilson et al.
Hum. Mol. Genet. 25 (5): 1031-1041. Code on github |
| November 2014 |
Extreme-depth re-sequencing of mitochondrial DNA finds no evidence of
paternal transmission in humans. Pyle et al. 2015. PloS Genetics. Data & Code |
| A Markov chain Monte Carlo strategy for sampling from the joint posterior distribution of pedigrees and population
parameters under a Fisher–Wright model with partial selfing. 2007. Theor. Popul. Biol.
The program described in this paper is Inference for Maternal Pedigrees with Partial Selfing IMPPS Home Page |
|
| QTLtree R library and stand alone program Home Page |
|
|
Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. 2014. Pfeffer et al. Code and data Shared Haplotypes. |
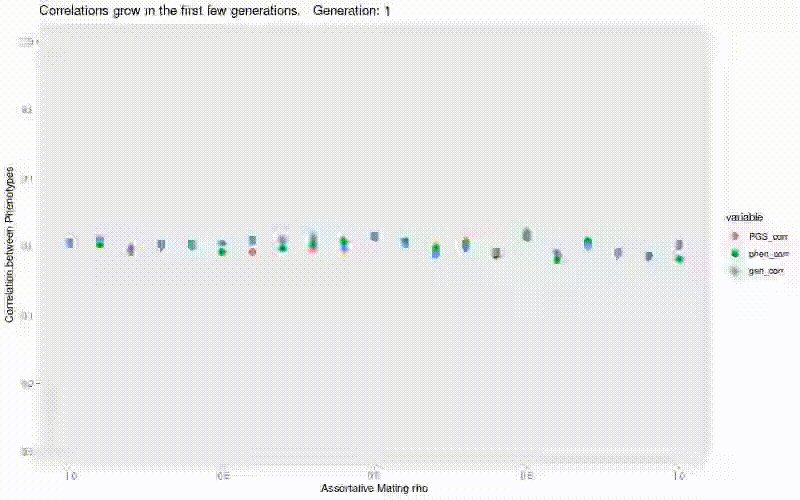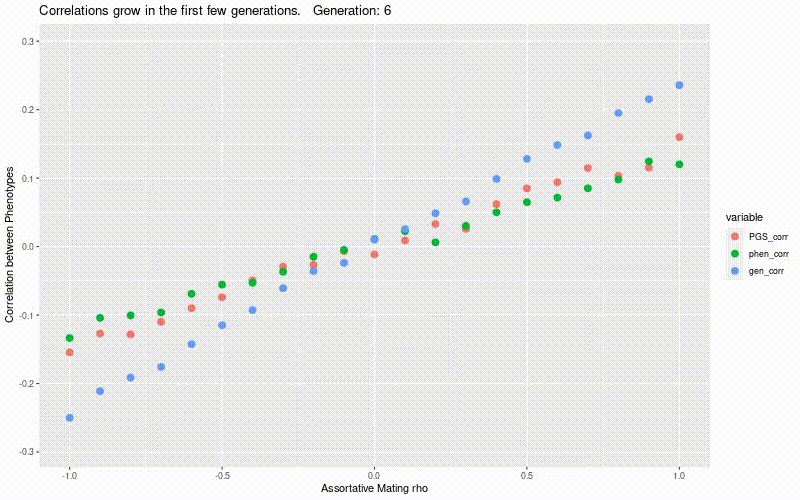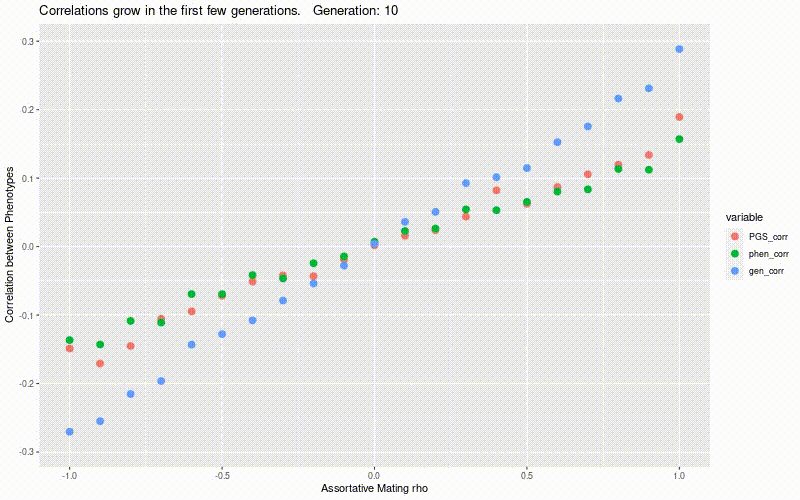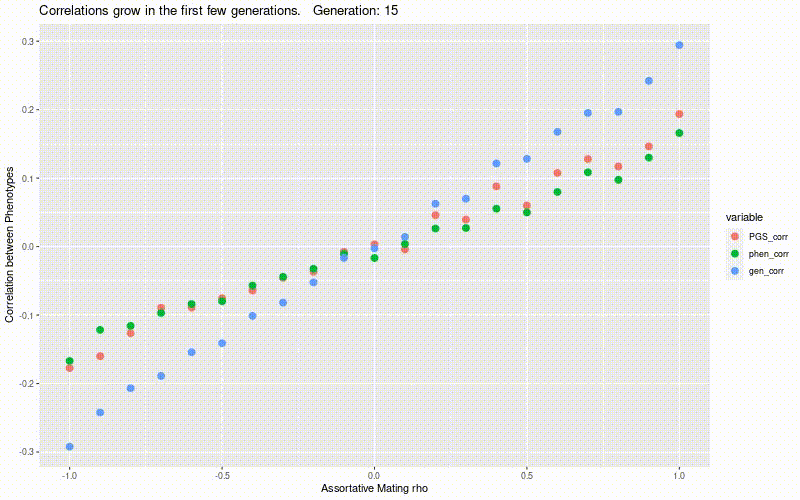
Simulate genetic correlation induced by assortative mating.
Packages (with varying degree of completeness) for the
R software system. Latest versions
in my R package directory
Install packages on unix/linux using the commands
R CMD INSTALL packagename.tar.gz
To install on a windows systems use the zip versions
and install from within R using
install.packages("packagename.zip",repos=NULL)
Where you have downloaded the package to your working directory. Once installed the
zip file can be safely deleted.
| genealogy.tar.gz | genealogy.zip | A package for the simulation of simple population genetic models |
| genomic.tar.gz | genomic.zip | A package for downloading and using HAPMAP and Perlegen datasets |
| pac.tar.gz | pac.zip | A package using Li & Stephens (2003) model for the analysis of genetic data, and my extensions for the analysis of data from subdivided populations. |
| ARG.tar.gz | ARG.zip | Simulate and extract information from Ancestral Recombination Graphs. Requires the ape package for plots. |
| batwing.tar.gz | batwing.zip | Package for postprocessing of BATWING output. |
| ijwtools.tar.gz | ijwtools.zip | My own set of utility functions (mainly for dealing with genetic data and the results from my own programs) including quick reading from xml files, omnibus tests, Manhattan distances between rows in a table, multivariate D' calculation. |
| snptree | Create and plot lexical trees from SNP data. Used in paper Recent mitochondrial DNA mutations increase the risk of developing multiple common late-onset human diseases, by Hudson et al. link |
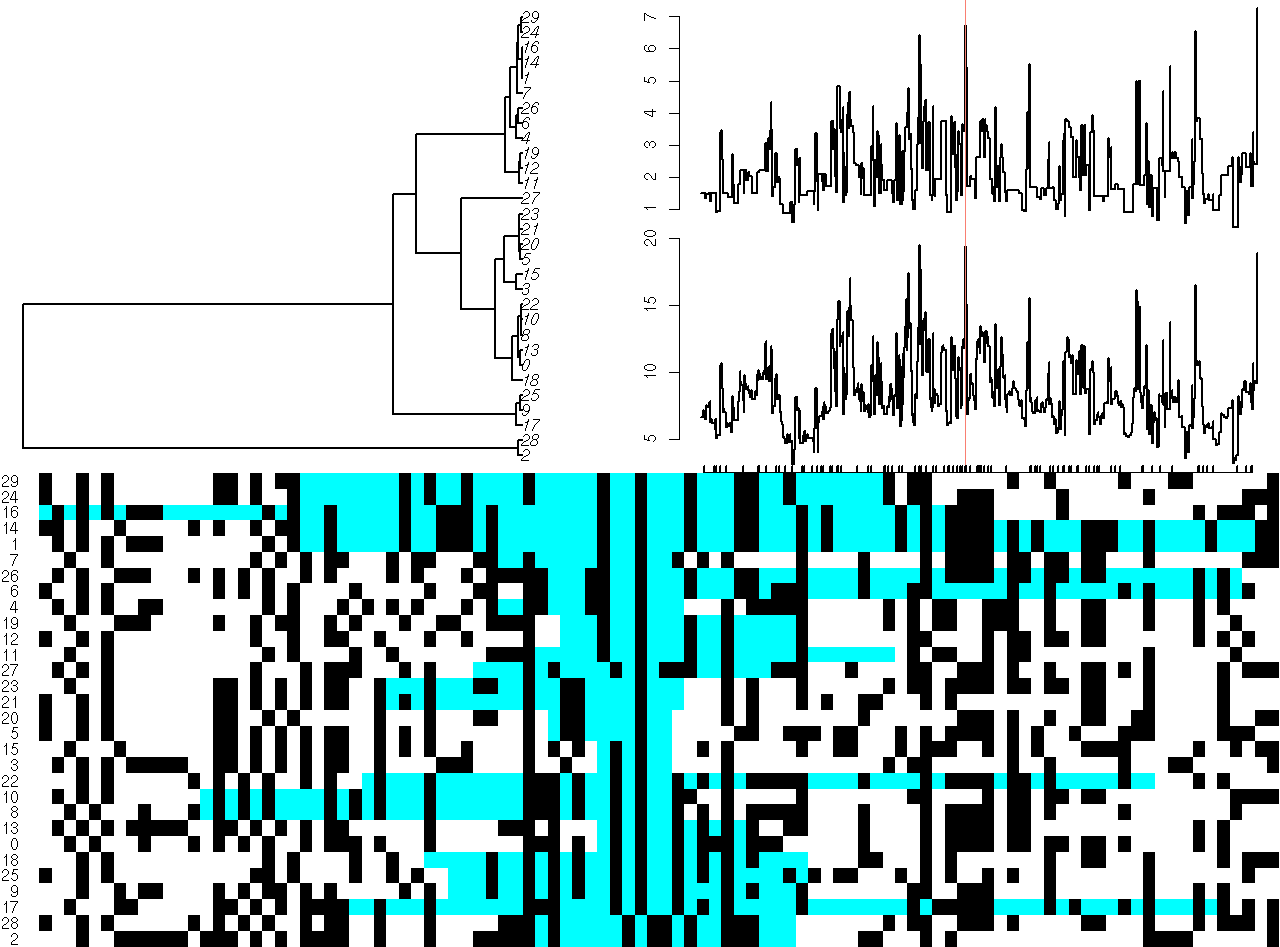
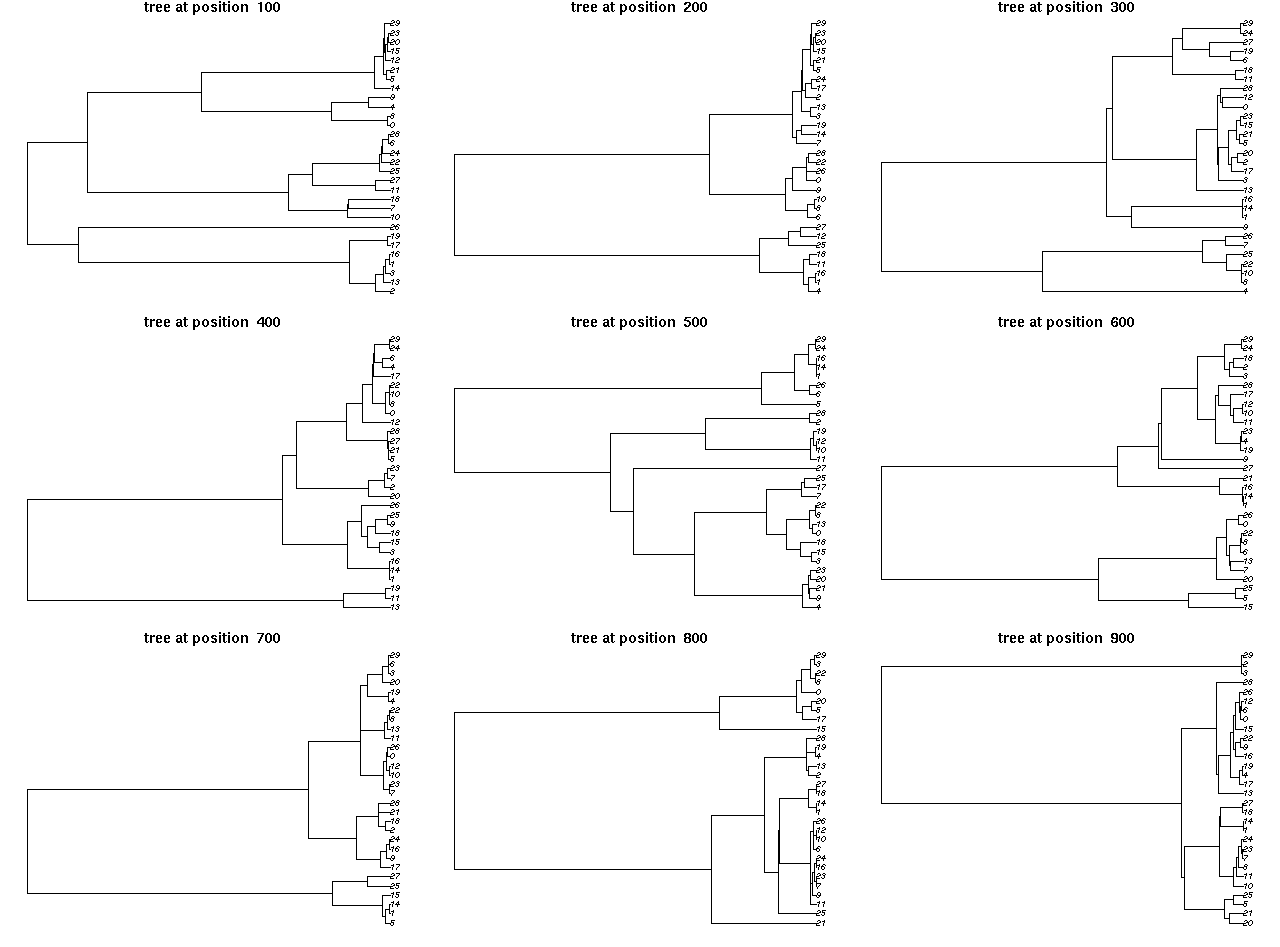
Screenshots of interactive plots from the ARG package. Left plot shows a view of an ancestral graph showing genetic variants (bottom panel) along with the tree under a single site (top left panel) and tree heights and lengths (top right panel). Right plot gives views of trees 100 sites apart. Click on either plot for a larger view.
All Software in this section is in the form of zipped tar files.
Use
gunzip -c filename.tar.gz | tar xf -
then type make
to compile the program. A limited manual for micsat is in the file
micsat.doc. Other manuals can be downloaded in pdf format.
The programs are distributed as executable files in a zip file format
A binhex-ed executable for the Macintosh, MacBatwing, may be downloaded.
On launching, the program opens a dialog box with a text field. The
user puts the program arguments (infile outfile seed) into this
field and hits ok.
(Compilation on the Mac courtesy of Oliver Pybus - send
comments/bugs to Ian.Wilson@ncl.ac.uk).
| Micsat | Batwing | Parentage | Hapgen | Ancestors | |
| Unix | micsat.tar.gz | batwing.tar.gz | parentage.tar.gz | hapgen.tar.gz | ancestors.tar.gz |
| Windows 95/NT | micsat.zip | batwing.exe | parentage.exe | ||
| Macintosh (MacOS 8.0 or above) | MacBatwing | ||||
| Manuals (pdf format) | micsat | bat_guide.pdf | parentage.pdf | ||
| Examples (inc R code) | parexamples.tar.gz | ||||
| Postprocessing (S/R code) | batwing.r | ||||
| Web interfaces | Web inferface at BioHP.org |
 |
MicsatThis is the program described in the paper "Genealogical inference from microsatellite data" by Wilson and Balding published in Genetics 150: 499-510. A description of the program and the downloaded files. |
|
|
Bayesian Analysis of Trees With Internal
Node Generation. Batwing is described in
Wilson, Weale & Balding 2003.Inferences from DNA data:
population histories, evolutionary processes and forensic match
probabilities. Journal of the Royal Statistical Society: Series
A, 166: 155-188. |
 |
Parentage
Program described in Emery, Wilson, Craig, Boyle and Noble (2001). Assignment of paternity groups without access to parental genotypes: multiple mating and development plasticity in squid. Molecular Ecology: 10: 1265-1278. downloadThe Computational Biology Service Unit at Cornell University provides free world-wide access to parentage via a web interface (BioHPC.org). Using this interface, researchers can run a variety of computationally intensive bioinformatics and population genetics tools, Parentage being one of them. |
A program for haplotype reconstruction from genotypes. Work done with David Cooper.
This program is a Markov chain Monte Carlo method to reconstruct haplotypes from genotypes. It uses Metropolis coupled chains to speed up mixing.
Includes options to deal with:
WinBUGS analysis of Twins Multivariate Normal Model, and Twins Stem Cell Number
Last Modified 18 June 2024